HPC 入门教程
本教程以 gromacs 软件为例,介绍如何从零开始在超算平台上运行计算任务及相关命令说明。
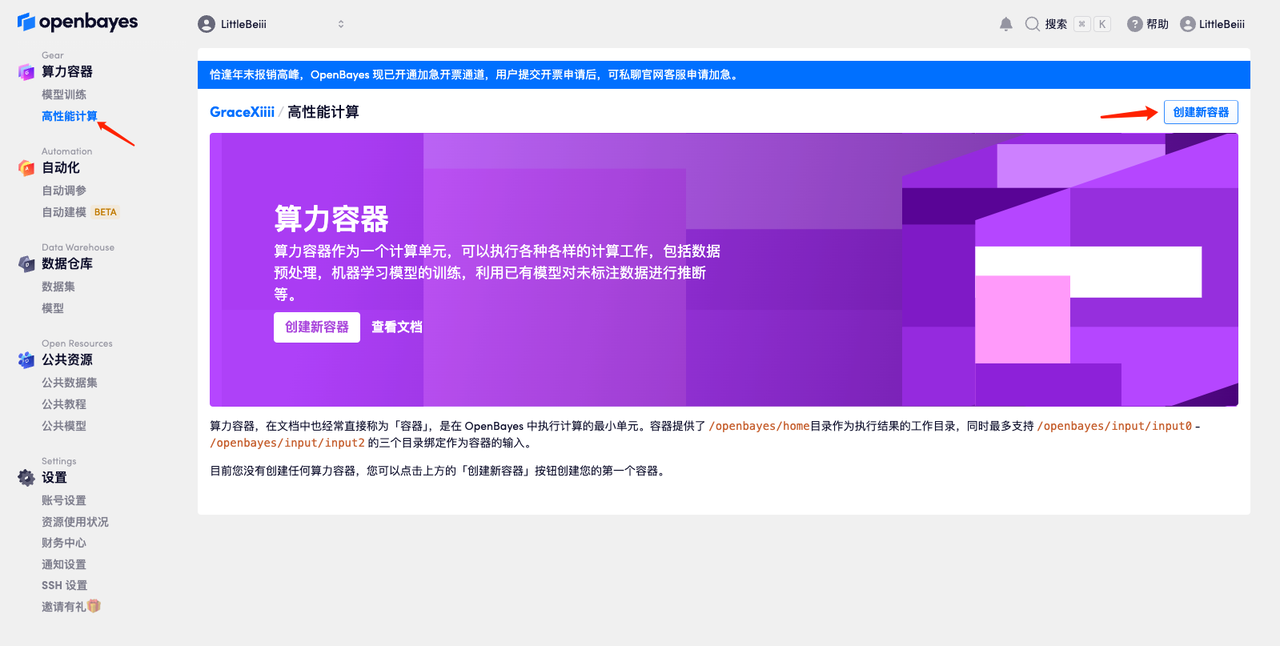
创建容器
- 点击左侧「高性能计算」,新建一个容器
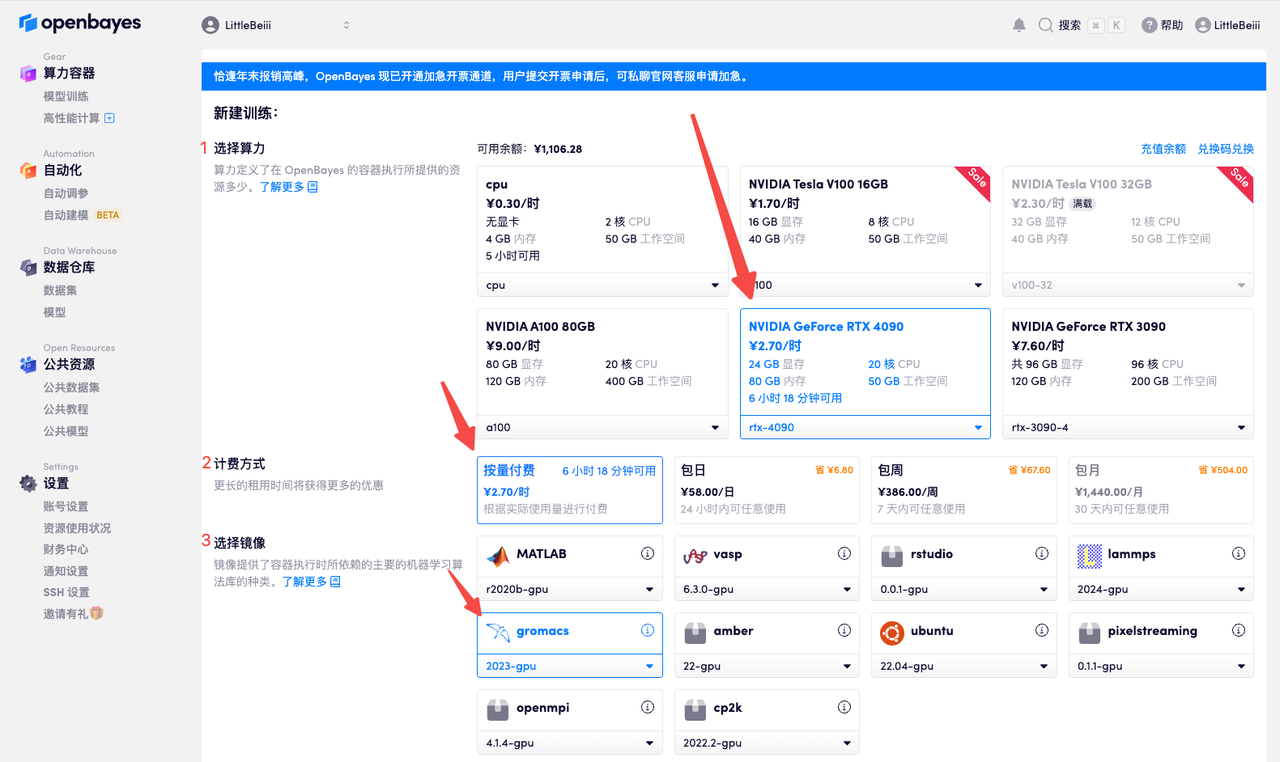
- 选择所需算力
- 「计费方式」处选择所需付费方式
- 「选择镜像」处选择所需软件,此处以「gromacs」为例
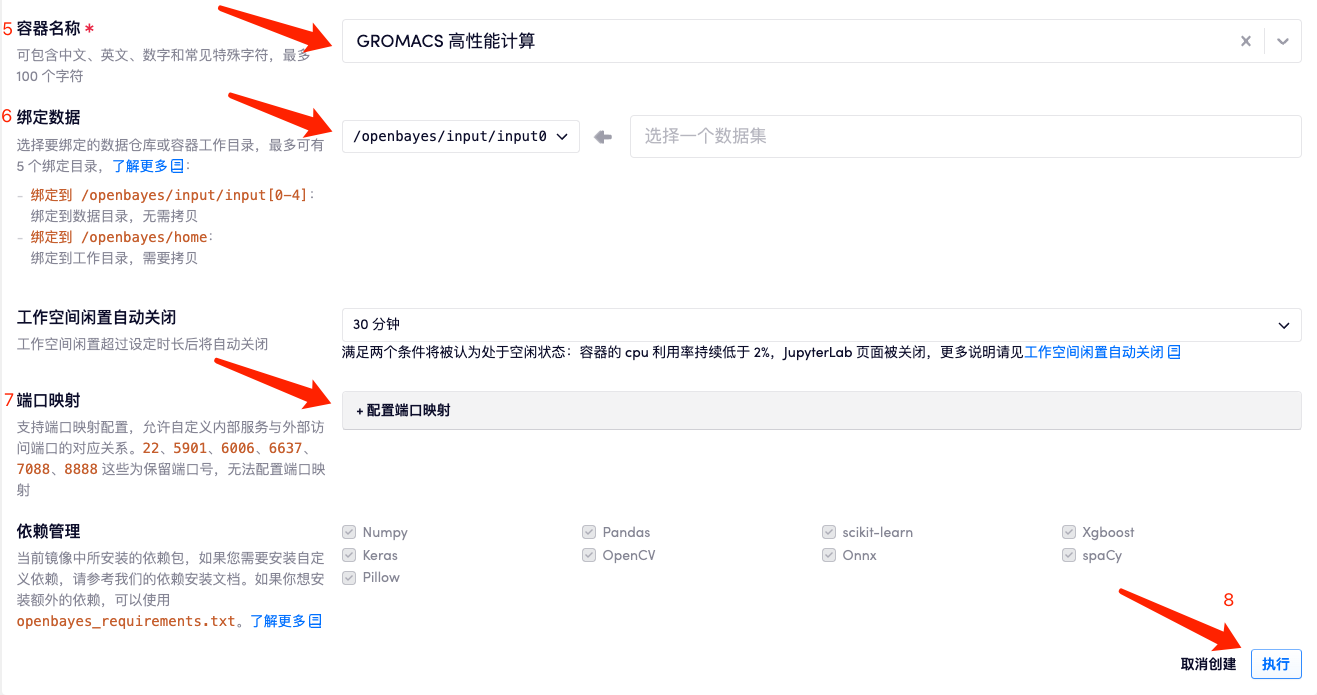
- 「容器名称」处输入 1 个合法的容器名称
- 「数据绑定」处选择要绑定的数据仓库或执行
- 根据自身需求,配置端口映射
- 点击「执行」
- 等待容器分配好资源,状态变为「运行中」后,点击「打开工作空间」。
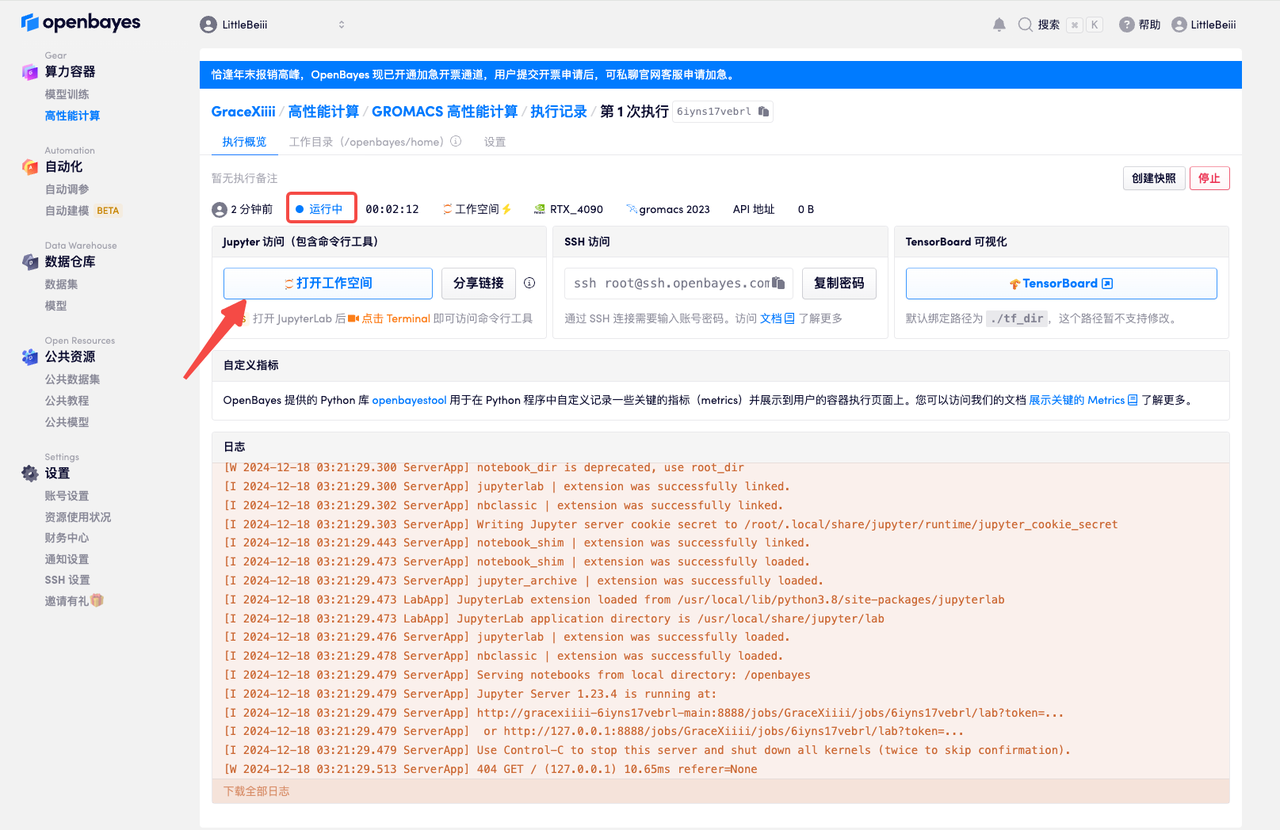
命令行运行 gromacs 任务
本案例将展示如何运用 gromacs 执行分子动力学模拟计算,完整任务运行约需 10h,此处仅提供相关命令说明。
准备输入文件及脚本
进行分子动力学计算时,需要准备以下 6 个关键文件(可在公共资源下载使用或直接 input):
laki.pdb(蛋白)、ions.mdp、md.mdp、minim.mdp、npt.mdp、nvt.mdp
(mdp 文件定义了分子动力学模拟的参数,包含了一系列的选项,用于控制模拟的各个方面,如时间步长、温度耦合、压力耦合、模拟类型)
上传输入文件
- 方法一:提前上传到自己的数据集,启动容器时直接 input,详情可参考 算力容器数据绑定
- 方法二:容器启动后直接在「工作空间」内上传或直接将文件拖入左侧对应目录处,如下图所示
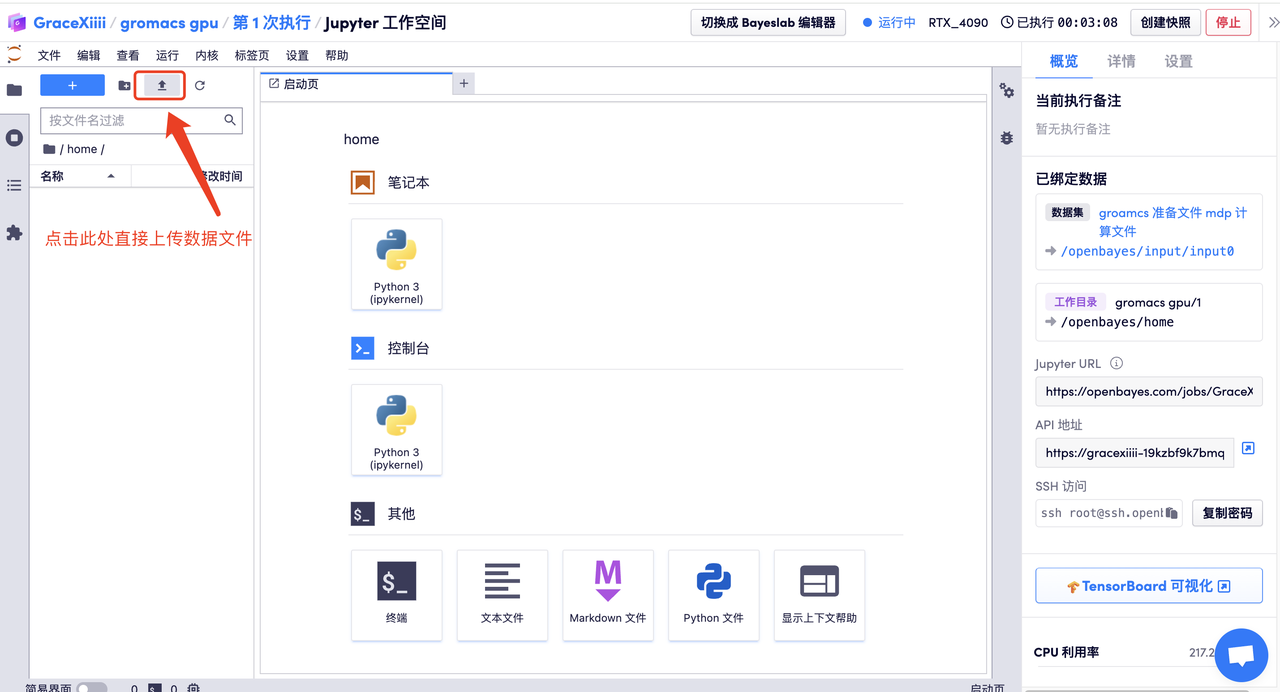
用 vim 创建文本文件
文本文件可以用 vim 创建文本文件到工作目录,例如在 input0 绑定的数据集「gromacs 准备文件 mdp 计算文件」创建文本文件:
打开一个终端,首先切换至自己的数据集目录下,这里切换 input0 目录:
cd /openbayes/input/input0
在终端输入 vim 即可进入 vim 编辑器,如下所示:
(base) root@liangzhong-4ay9ej85pxvd-main:/openbayes/input/input0# ls
1aki.pdb
(base) root@liangzhong-4ay9ej85pxvd-main:/openbayes/input/input0# vim test.txt
Vim 常用代码:
- 输入 i 进入插入模式
- 输入 Esc 退出插入模式
- 输入 :wq 保存退出
使用软件
软件调用
以 gromacs 为例,配置环境变量,其他软件只需替换此命令对应的路径「/data/app/gromacs/bin」:
export PATH=/data/app/gromacs/bin:$PATH
提交任务命令
使用 nohup 确保 gromacs 的 mdrun 命令在后台运行。它将使用 GPU 进行邻居搜索,使用 CPU 进行 PME 计算,并将所有输出保存到「log.txt」文件中:
nohup gmx_mpi mdrun -deffnm md_0_1 -v -nb gpu -pme cpu > log.txt &
脚本说明:
- 输入该代码后将开始计算任务。为避免在运行任务时,终端会实时弹出运行日志,无法进行其他操作,可以在代码后添加「> log.txt &」使得日志报告不再于终端中弹出,而是自动写入「log.txt」的文本。
gmx_mpi mdrun -deffnm md_0_1 -v -nb gpu -pme cpu可以替换为其他命令,如 bash、sh、py 等执行文件。以下为上述命令具体说明:nohup:该命令会忽略所有的挂起信号,即使用户退出终端会话,指定的命令仍然会继续执行。gmx_mpi mdrun:这是 gromacs 软件中用于执行分子动力学模拟的 MPI 版本命令。gmx_mpi 是 gromacs 的 MPI 版本,用于在多处理器系统上并行运行模拟。-deffnm md_0_1:这个选项指定了模拟的默认输出文件名。在这里,md_0_1 是输出文件的基本名称,gromacs 会根据这个名称生成一系列的输出文件,如能量文件、日志文件等。-v:这个选项使 mdrun 命令在运行时输出详细的信息,包括进度、性能统计等。-nb gpu:这个选项指定了邻居搜索算法应该在 GPU 上执行。nb 代表「neighbor 搜寻」,这是分子动力学模拟中的一个关键步骤,用于确定原子之间的相互作用。-pme cpu:这个选项指定了 PME(粒子网格 Ewald)算法,用于处理长程电荷相互作用,应该在 CPU 上执行。PME 是一种常用的方法,用于在保持周期性边界条件的同时,高效地计算电荷相互作用。log.txt:这部分将命令的标准输出重定向到文件 log.txt。这意味着所有由 mdrun 命令打印到控制台的输出都会被保存在这个文件中。&:这个符号表示命令将在后台运行。即使用户退出终端会话,命令仍然会继续执行。
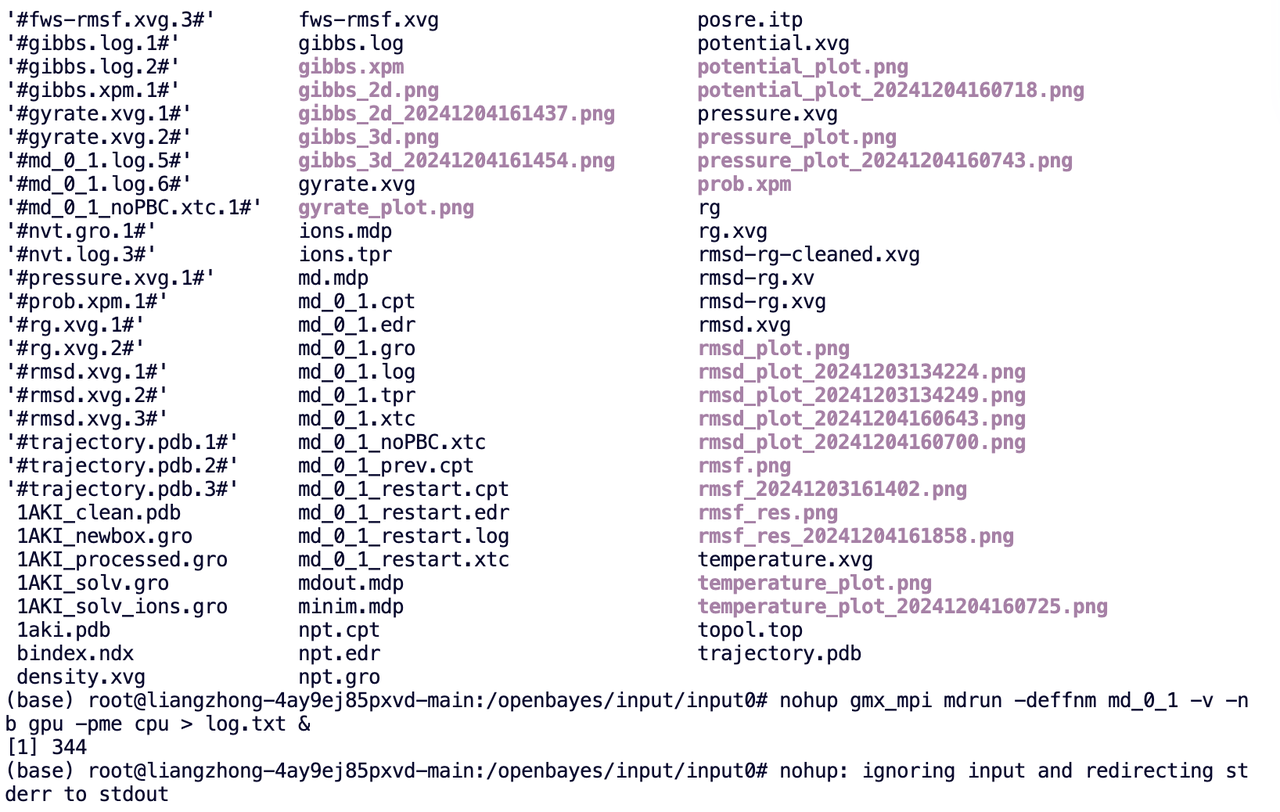
算力任务(作业)状态查看
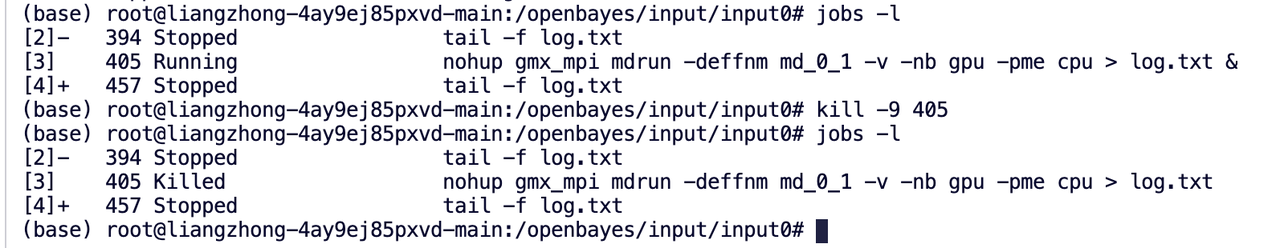
命令行查看作业状态命令:
jobs -l

使用 tail 可以查看实时输出:
tail -f log.txt
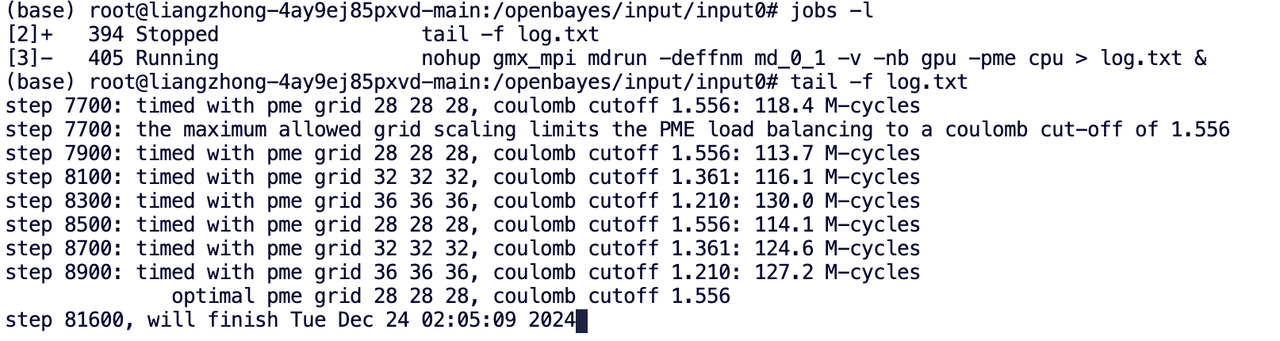
tail 是一个读取文本的末端的阅读器,可以根据「log.txt」的实时更新,不断弹出新的内容。输入 ctrl + c 可退出「log.txt」文本。
如果要取消作业,可用命令:
kill -9 进程id
计算结果文件下载
当计算任务运行完毕后,可以关闭容器,避免计费。
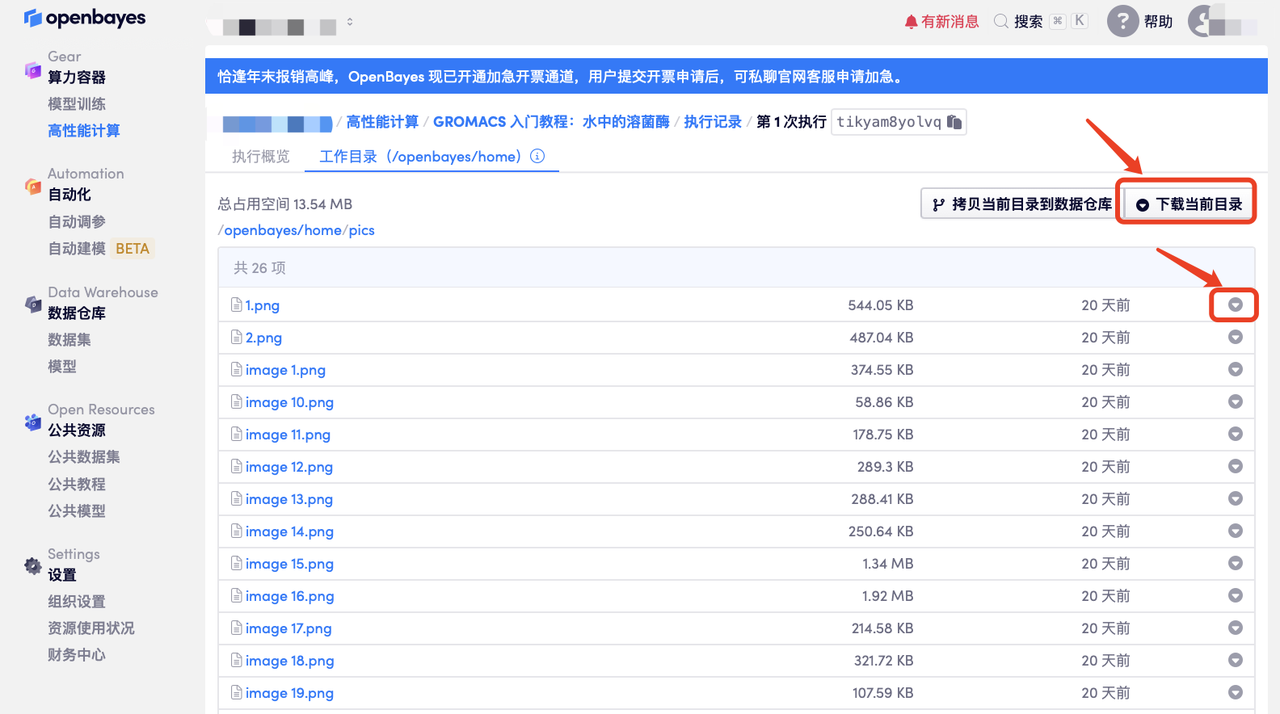
「打开容器」-「执行记录」-「工作空间目录」-「选择需要下载的文件」即可下载结果文件到本地,参考如下图:
此处可以选择「下载当前目录」或直接下载单个文件。
如果结果是保存在之前 input 的数据集中,可以直接在「数据仓库」中打开对应数据集:
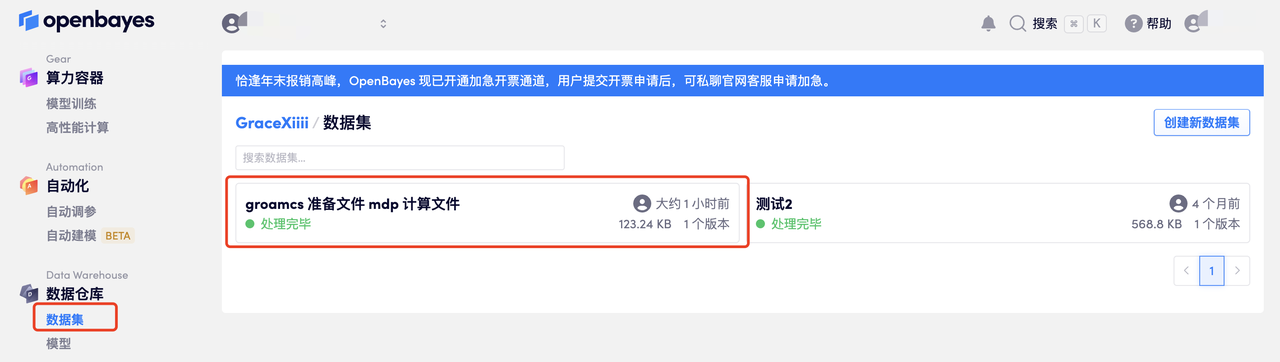
这里可以看到自己的数据,点击「下载当前目录」即可获取当前目录下的所有文件到本地电脑:
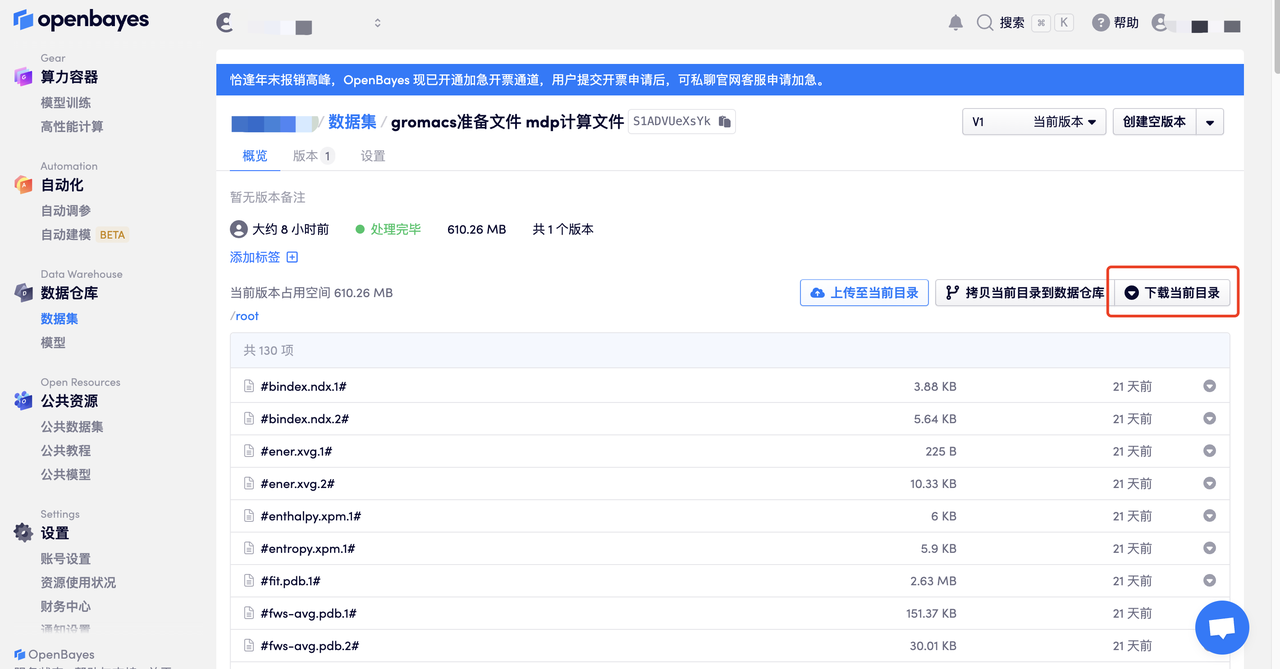
或选择你需要的某个文件,下载当前文件/在线查看 .png 等图片文件。
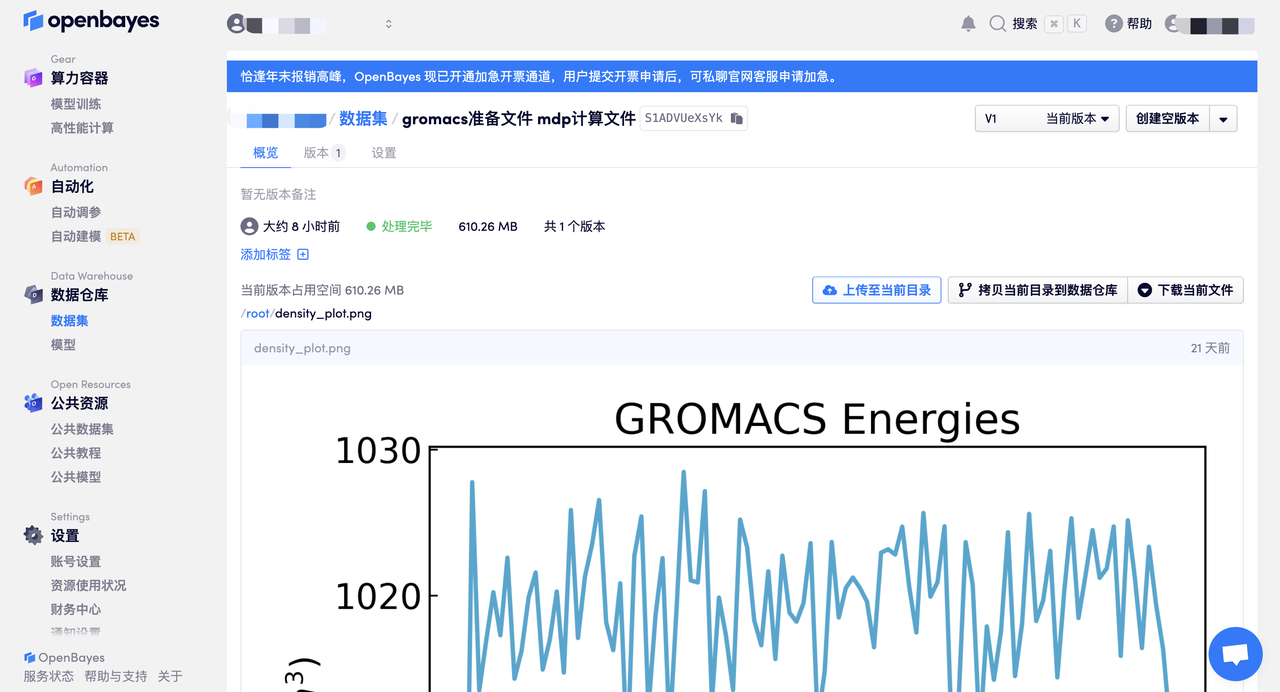
gromacs 任务完成后直接下载生成的图片和数据即可。
远程 SSH 连接
这里介绍如何使用远程 SSH 连接到本地。
在工作空间创建成功后,页面会出现 SSH 登录相关的信息,也会为该容器创建一个密码,通过该密码可以完成 SSH 的身份验证,无需其他步骤。
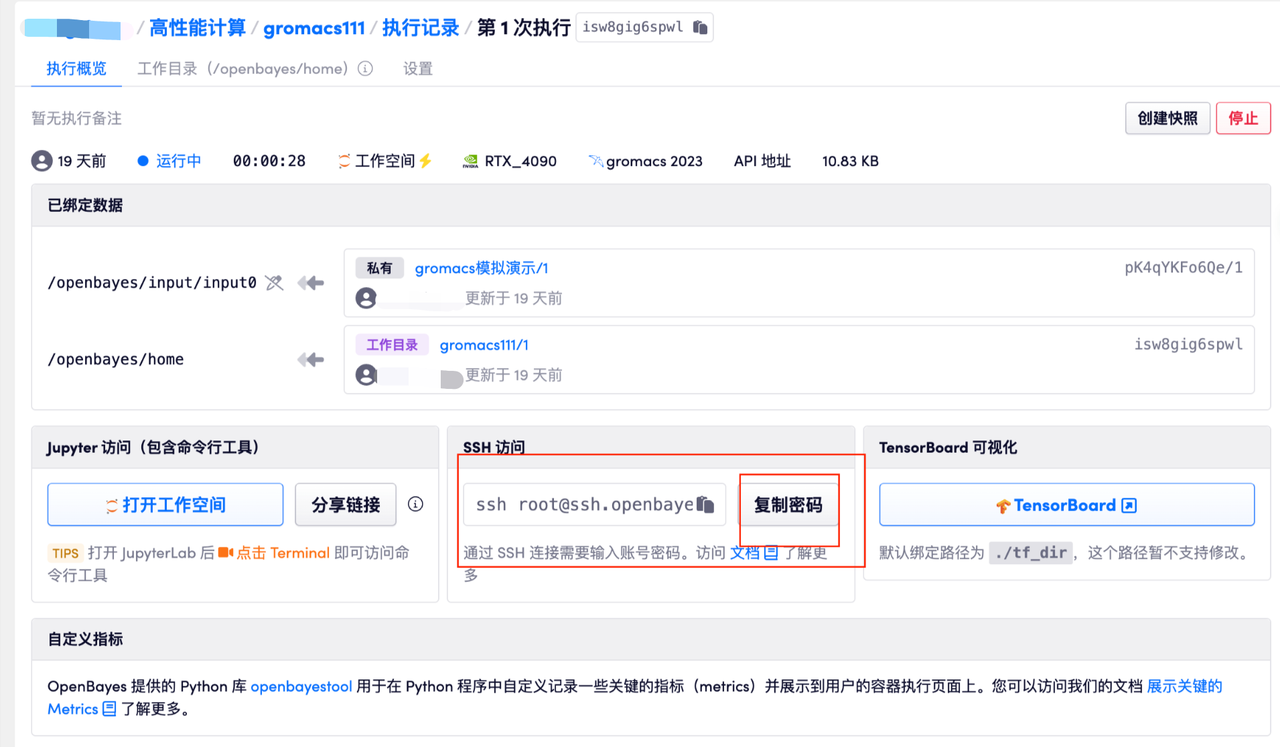
方法一:在支持 SSH 的客户端中连接服务器
详细文档见通过 SSH 连接运行中的 Jupyter 工作空间。
方法二:X-shell(需要使用自己的邮箱注册下载正版软件)
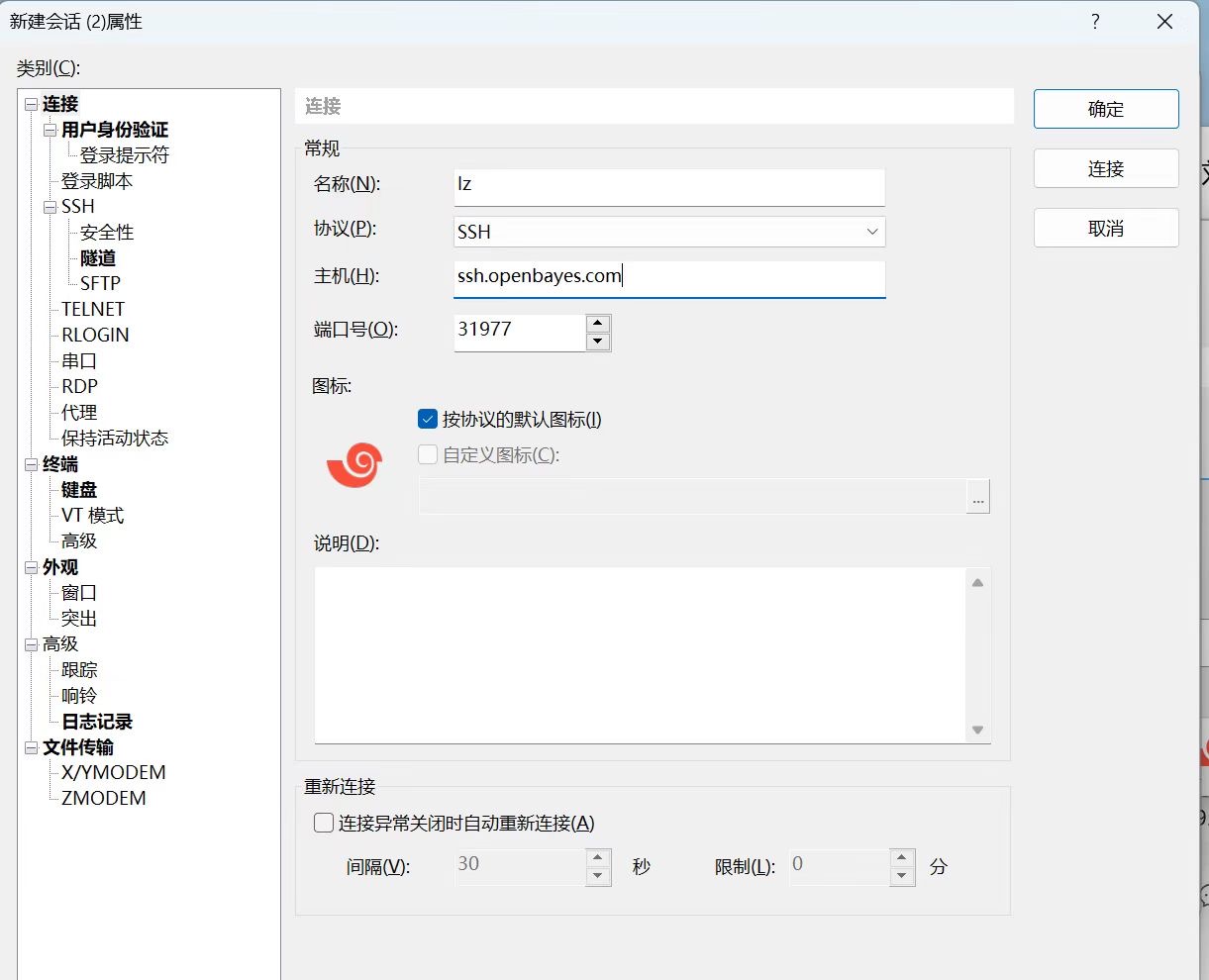
在本地打开 X-shell,新建 SSH 连接(如下图所示):
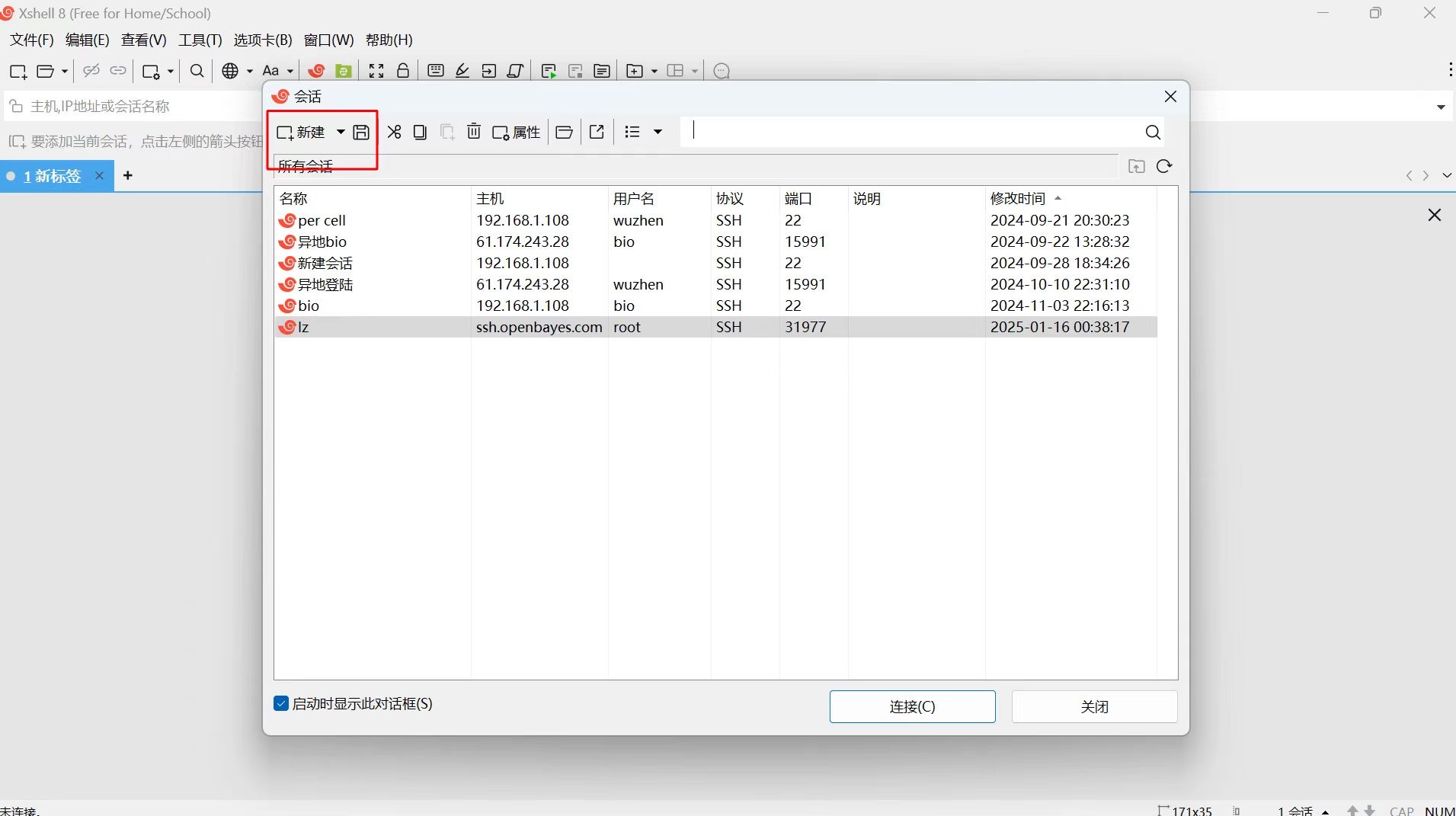
在链接界面输入以下平台容器信息:
- 名称:(这里输入 1 个合法的名称)
- 协议:SSH
- 主机:openbayes.com
- 端口号:31977(这里需要输入所启动容器的对应端口号)
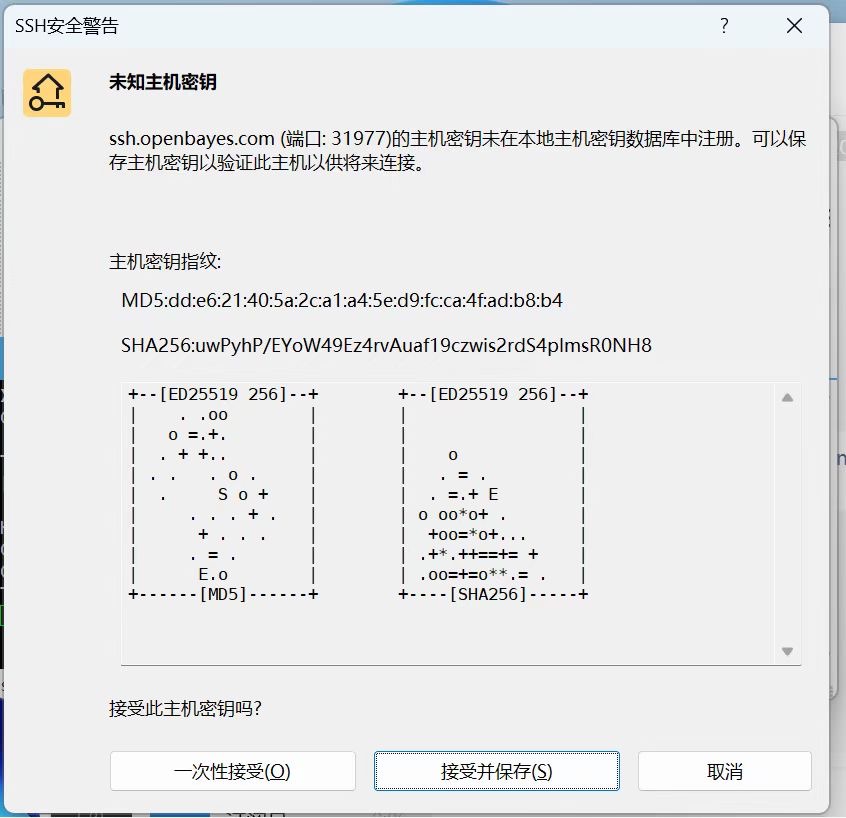
接受主机密钥
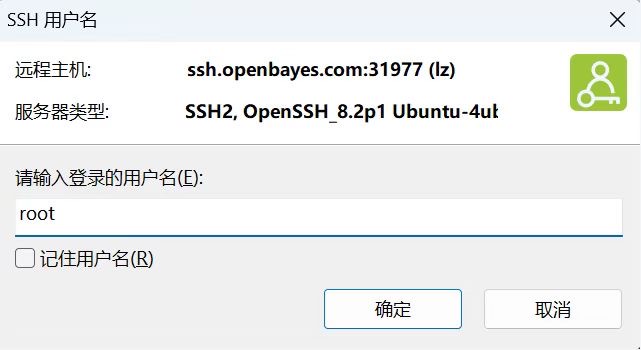
输入用户名:root 输入自定义密码
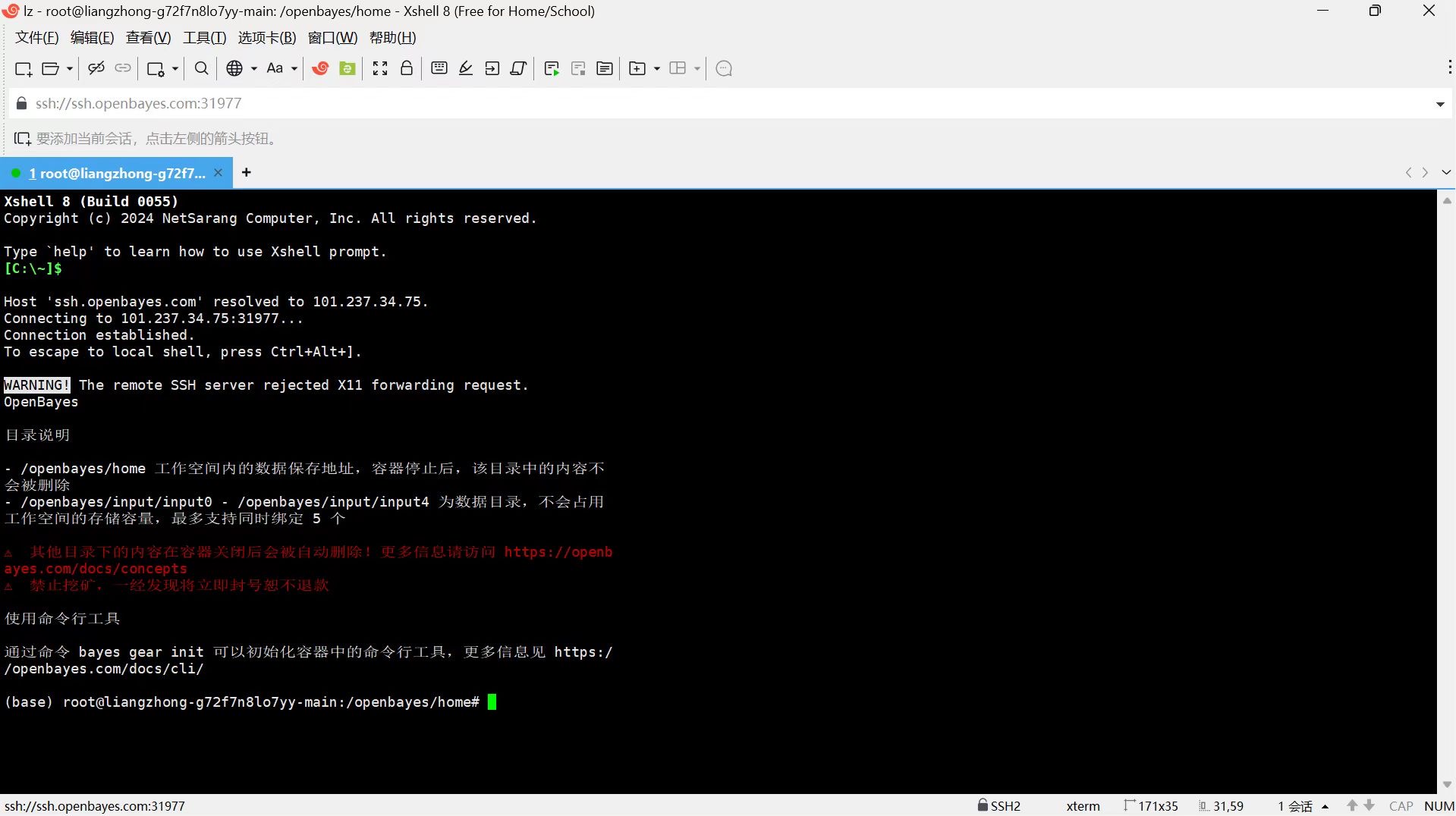
连接成功(如下图所示):
常见问题
软件位置在哪里?
软件 app 的位置在 data/app 目录下。

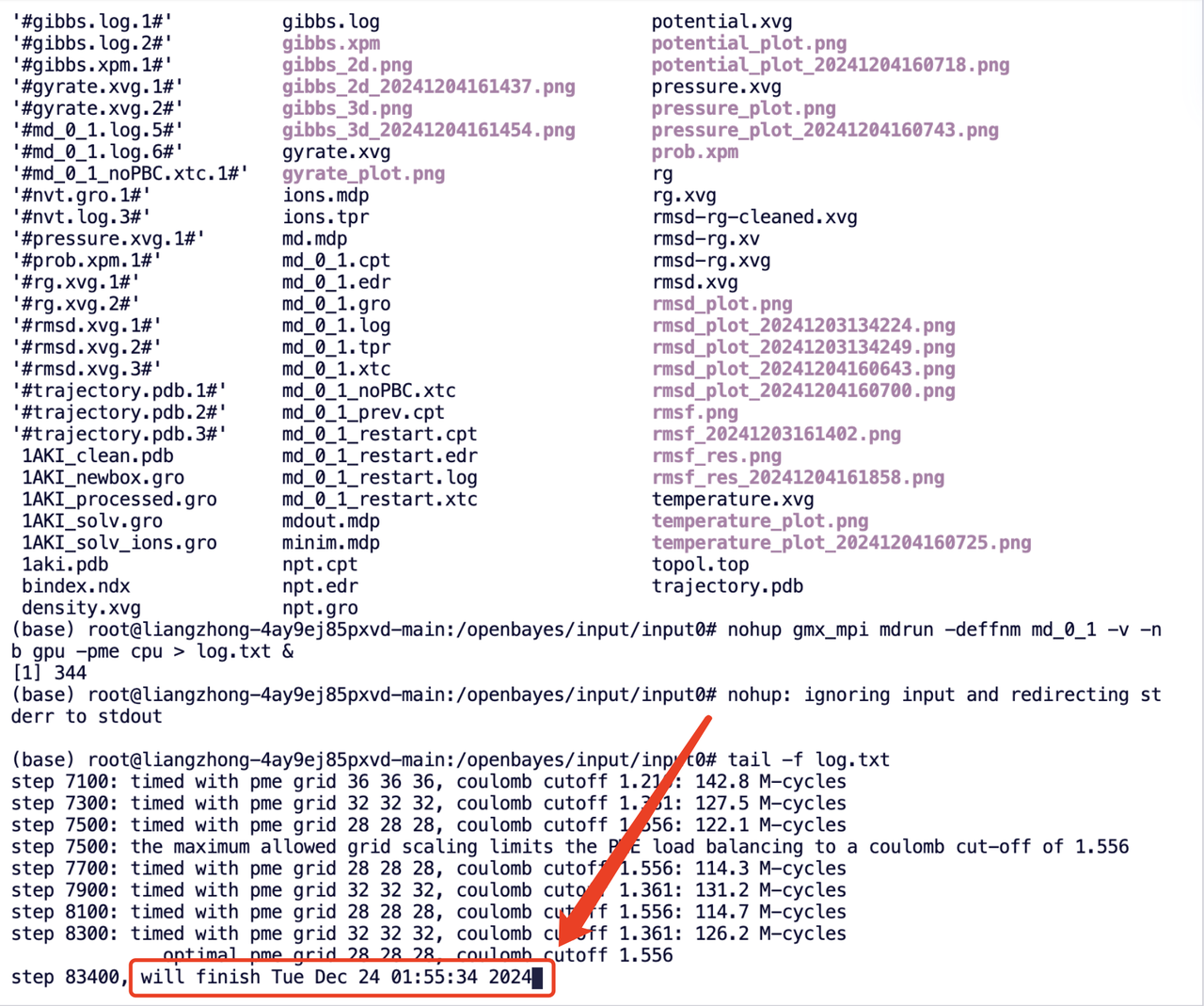
容器内时区是什么?
容器里默认展示的是 UTC 时间,即 0 时区时间,所以服务器中提供的算力任务预计完成时间需要加 8 小时为北京时间。
例如:下图中显示该作业任务将于 2024 年 12 月 24 日 1:55:34 完成,所以实际完成时间约为北京时间 2024 年 12 月 24 日 9:55:34。